Ung thư
Stivarga

Hoạt chất:
Mỗi viên nén bao phim chứa 40 mg regorafenib.
Các tá dược:
Lõi viên không bao: Cellulose vi tinh thể, Croscarmellose natri, Magnesi stearat, Povidon, Silica colloidal khan
Màng bao phim: Oxyd sắt đỏ, Oxyd sắt vàng, Lecithin (xuất nguồn từ đậu nành), Macrogol, cồn Polyvinyl thủy phân một phần, bột Talc, Titan dioxyd
Viên nén bao phim.
Viên nén bao phim màu hồng nhạt, hình bầu dục với chiều dài 16 mm và chiều rộng 7 mm với chữ 'BAYER’ được dập nổi ở một bên và '40' ở phía bên kia.
Stivarga được chỉ định để điều trị cho những bệnh nhân ung thư đại trực tràng (colorectal cancer - CRC) di căn trước đó đã được điều trị bằng, hoặc không được coi là ứng cử viên để sử dụng phác đồ hóa trị có dẫn xuất fluoropyrimidin, liệu pháp kháng VEGF, và một liệu pháp kháng EGFR nếu bệnh nhân có tuýp RAS hoang dã.
Stivarga được chỉ định để điều trị cho các bệnh nhân có khối u mô đệm đường tiêu hóa (gastrointestinal stromal tumors - GIST) không còn khả năng phẫu thuật hoặc di căn có tiến triển hoặc không dung nạp với phác đồ điều trị trước đó với imatinib và sunitinib.
Stivarga được chỉ định để điều trị cho bệnh nhân bị ung thư biểu mô tế bào gan (hepatocellular carcinoma - HCC) đã được điều trị trước đó bằng sorafenib.
Stivarga phải được kê đơn bởi bác sĩ có kinh nghiệm sử dụng hóa trị liệu điều trị ung thư.
Cách dùng
Dùng đường uống
Chế độ liều
Liều khuyến cáo là 160 mg regorafenib (4 viên Stivarga, mỗi viên chứa 40 mg regorafenib), uống một lần mỗi ngày trong 3 tuần điều trị, sau đó nghỉ điều trị 1 tuần để tạo thành một chu kỳ 4 tuần.
Stivarga nên được uống cùng thời điểm mỗi ngày. Các viên thuốc phải được nuốt nguyên viên với nước sau một bữa ăn nhẹ. Nếu quên một liều Stivarga, sau đó nên uống lại trong cùng một ngày ngay sau khi bệnh nhân nhớ ra. Bệnh nhân không nên dùng hai liều trong cùng một ngày để bù đắp cho liều đã quên. Trong trường hợp bệnh nhân nôn sau khi uống regorafenib, không nên dùng thêm viên nữa.
Nên tiếp tục điều trị khi vẫn thấy có lợi ích hoặc cho đến khi xảy ra độc tính không thể chấp nhận được (xem phần 'Cảnh báo đặc biệt và thận trọng').
Bệnh nhân có chỉ số toàn trạng (PS) bằng 2 hoăc cao hơn được loại ra khỏi nghiên cứu lâm sàng. Có rất ít dữ liệu trên bệnh nhân có chỉ số PS¬≥2.
Hiệu chỉnh liều
Có thể cần ngừng và / hoặc giảm liều dựa trên an toàn và khả năng dung nạp của từng cá thể. Hiệu chỉnh liều được áp dụng theo từng bậc 40 mg (một viên). Liều khuyến cáo hàng ngày thấp nhất là 80 mg. Liều dùng hàng ngày tối đa là 160 mg.
Để khuyến cáo hiệu chỉnh liều và các biện pháp áp dụng trong trường hợp có phản ứng da bàn tay-chân (Hand Foot Skin Reaction- HFSR), xem Bảng 1.
Bảng 1: Hiệu chỉnh liều và các biện pháp được khuyến cáo đối với phản ứng da bàn tay-chân (HFSR)
|
Mức độ độc tính trên da |
Xuất hiện |
Hiệu chỉnh liều và các biện pháp được khuyến cáo
|
|
Độ 1 |
Xuất hiện bất kỳ lần nào |
Duy trì liều dùng và áp dụng ngay các biện pháp hỗ trợ để làm giảm triệu chứng.
|
|
Độ 2 |
Xuất hiện lần đầu |
Liều giảm 40 mg (một viên) và áp dụng ngay các biện pháp hỗ trợ. |
|
|
Không cải thiện trong vòng 7 ngày hoặc xuất hiện lần 2
|
Tạm ngừng điều trị cho đến khi độc tính được giải quyết về độ 0-1. |
|
|
Xuất hiện lần thứ 3 |
Tạm ngừng điều trị cho đến khi độc tính được giải quyết về độ 0-1. |
|
|
Xuất hiện lần thứ 4 |
Dừng điều trị |
|
Độ 3 |
Xuất hiện lần đầu |
Áp dụng ngay các biện pháp hỗ trợ. Tạm ngừng điều trị ít nhất 7 ngày cho đến khi độc tính được giải quyết về độ 0-1. Khi điều trị lại, giảm liều 40mg (1 viên). Việc tăng một mức liều là được phép theo quyết định của bác sĩ điều trị. |
|
|
Xuất hiện lần thứ 2 |
Áp dụng ngay các biện pháp hỗ trợ. Tạm ngừng điều trị ít nhất 7 ngày cho đến khi độc tính được giải quyết về độ 0-1. Khi điều trị lại, giảm liều 40mg (1 viên). |
|
|
Xuất hiện lần thứ 3 |
Ngừng điều trị |
Các cách hiệu chỉnh liều và các biện pháp cần áp dụng được khuyến cáo trong trường hợp các xét nghiệm chức năng gan xấu đi có liên quan đến trị liệu bằng Stivarga, xem bảng 2 (cũng xem phần 'Cảnh báo đặc biệt và thận trọng’).
Bảng 2: Các biện pháp và cách hiệu chỉnh liều được khuyến cáo trong trường hợp có bất thường về các xét nghiệm chức năng gan liên quan đến thuốc
|
Tăng ALT và/ hoặc AST |
Xuất hiện |
Các biện pháp và cách hiệu chỉnh liều được khuyến cáo
|
|
≤ 5 lần giới hạn trên bình thường (ULN) (tối đa độ 2) |
Lần xuất hiện bất kỳ |
Tiếp tục điều trị Stivarga. |
|
> 5 lần ULN đến ≤ 20 lần ULN (độ 3) |
Lần xuất hiện đầu tiên |
Tạm ngừng điều trị Stivarga. |
|
|
|
Bắt đầu dùng lại: nếu lợi ích lớn hơn nguy cơ độc tính trên gan, khởi đầu lại điều trị với Stivarga, giảm liều đi 40mg (1 viên) và giám sát chức năng gan hàng tuần trong ít nhất 4 tuần. |
|
|
Tái xuất hiện |
Ngừng hẳn việc điều trị bằng Stivarga. |
|
> 20 lần ULN |
Lần xuất hiện bất kỳ |
Ngừng hẳn việc điều trị bằng Stivarga.
|
|
> 3 lần ULN ( độ 2 hoặc cao hơn) kèm theo bilirubin > 2 lần ULN |
Lần xuất hiện bất kỳ |
Ngừng hẳn việc điều trị bằng Stivarga. Giám sát chức năng gan hàng tuần cho đến khi giải quyết hoặc trở về mức trước điều trị. Ngoại lệ: Các bệnh nhân bị hội chứng Gilbert’s có tăng các transaminase cần được điều trị như khuyến cáo ở trên đối với riêng dấu hiệu tăng ALT và/hoặc AST. |
Thông tin bổ sung về các đối tượng bệnh nhân đặc biệt
Các bệnh nhân suy gan
Regorafenib được thải trừ chủ yếu qua gan
Trong các nghiên cứu lâm sàng, không quan sát thấy có sự khác biệt lâm sàng liên quan về sự phơi nhiễm, an toàn và hiệu quả giữa các bệnh nhân có suy gan nhẹ (Child-Pugh A) so với các bệnh nhân có chức năng gan bình thường. Không cần điều chỉnh liều ở bệnh nhân suy gan nhẹ. Bởi vì dữ liệu trên bệnh nhân suy gan trung bình (Child Pugh B) rất hạn chế, không có liều khuyến cáo cho bệnh nhân này.
Khuyến cáo giám sát chặt chẽ an toàn trên những bệnh nhân này (xem thêm phần "Cảnh báo đặc biệt và thận trọng khi sử dụng” và “Các đặc tính dược động học”).
Stivarga không được khuyến cáo sử dụng ở những bệnh nhân suy gan nặng (Child-Pugh C) vì Stivarga chưa được nghiên cứu trong nhóm dân số này.
Bệnh nhân suy thận:
Các dữ liệu lâm sàng đã có cho thấy sự tương tự về mức độ phơi nhiễm của regorafenib và các chất chuyển hóa của nó M-2 và M-5 trên những bệnh nhân suy nhận nhẹ, vừa và nặng so với những bệnh nhân có chức năng thận bình thường. Không cần điều chỉnh liều ở những bệnh nhân suy thận nhẹ hoặc trung bình (xem thêm phần ‘Các đặc tính dược động học’).
Người cao tuổi
Trong các nghiên cứu lâm sàng, không có sự khác nhau đáng kể trong phơi nhiễm, tính an toàn hoặc hiệu quả được quan sát giữa bệnh nhân cao tuổi (tuổi ≥65) và bệnh nhân trẻ. Có rất ít thông tin ở bệnh nhân trên 75 tuổi.
Trẻ em
Không dùng Stivarga ở bệnh nhi trong chỉ định ung thư đại trực tràng.Tính an toàn và hiệu quả của regorafenib ở bệnh nhân dưới 18 tuổi trong chỉ định GIST chưa được chứng minh. Chưa có dữ liệu.
Không dùng Stivarga ở bệnh nhi trong chỉ định ung thư biểu mô tế bào gan.
Sự khác biệt về chủng tộc
Trong các nghiên cứu lâm sàng, không quan sát thấy sự khác biệt có ý nghĩa về mức độ tiếp xúc, tính an toàn hoặc hiệu quả giữa các bệnh nhân của các nhóm chủng tộc khác nhau. Không cần điều chỉnh liều dựa trên chủng tộc (xem phần 'Các đặc tính dược động học'). Đã quan sát thấy một tỷ lệ cao hơn của phản ứng da bàn tay – bàn chân (hand foot skin reaction - HFSR), các bất thường nặng về xét nghiệm chức năng gan và rối loạn chức năng gan ở các bệnh nhân người Châu Á (đặc biệt ở Nhật Bản) được điều trị bằng Stivarga so với người da trắng. Các bệnh nhân người châu Á được điều trị với Stivarga trong các nghiên cứu lâm sàng chủ yếu là từ Đông Á (~ 90%).
Bệnh nhân quá mẫn với hoạt chất và bất kỳ thành phần tá dược nào trong công thức thuốc.
Các tác dụng trên gan
Những bất thường về xét nghiệm chức năng gan (alanine aminotransferase -ALT, aspartate aminotransferase -AST và bilirubin) thường được quan sát ở những bệnh nhân được điều trị với Stivarga. Những bất thường nghiêm trọng về các xét nghiệm chức năng gan (độ 3 đến 4) và rối loạn chức năng gan với các biểu hiện lâm sàng (bao gồm cả tử vong) đã được báo cáo trên một tỷ lệ nhỏ các bệnh nhân (xem phần ‘Tác dụng không mong muốn’). Trong các nghiên cứu lâm sàng, đã ghi nhận tỉ lệ cao hơn các bất thường về xét nghiệm chức năng gan và rối loạn chức năng gan nặng ở bệnh nhân châu Á (đặc biệt là Nhật Bản) khi điều trị với Stivarga, so với chủng người da trắng (xem phần Liều lượng và cách dùng).
Khuyến cáo thực hiện xét nghiệm chức năng gan (ALT, AST và bilirubin) trước khi bắt đầu điều trị Stivarga và giám sát chặt chẽ (ít nhất 2 tuần/lần) trong vòng 2 tháng đầu điều trị. Sau đó, nên tiếp tục giám sát định kỳ ít nhất là hàng tháng và khi được chỉ định lâm sàng.
Regorafenib là một chất ức chế men uridine diphosphate glucuronosyl transferase UGT1A1 (xem phần 'Tương tác với các thuốc khác và các dạng tương tác khác'). Tăng bilirubin gián tiếp (không liên hợp) nhẹ có thể xảy ra ở các bệnh nhân bị hội chứng Gilbert.
Đối với những bệnh nhân có xét nghiệm chức năng gan xấu đi có thể do điều trị bằng Stivarga (ví dụ khi không có nguyên nhân nào khác rõ ràng, như tắc mật sau gan hoặc tiến triển của bệnh), cần tuân thủ việc điều chỉnh liều và những lời khuyên về giám sát trong Bảng 2 (xem phần ‘Liều dùng và cách dùng’ – mục ‘Hiệu chỉnh liều’).
Cần giám sát chặt chẽ về an toàn chung trên các bệnh nhân có suy gan nhẹ hoặc trung bình (xem phần ‘Liều dùng và cách dùng’ – mục ‘Bệnh nhân suy gan’ và ‘Các đặc tính dược động học’). Stivarga không được khuyến cáo sử dụng ở những bệnh nhân suy gan nặng (Child-Pugh C) vì Stivarga chưa được nghiên cứu trong nhóm dân số này và sự phơi nhiễm có thể bị tăng lên ở những bệnh nhân này.
Nhiễm trùng
Stivarga có liên quan đến gia tăng tần suất của các biến cố nhiễm trùng, một số trong đó đã tử vong (xem phần 'Tác dụng không mong muốn').
Trong các trường hợp có biến cố nhiễm trùng ngày càng xấu đi, nên xem xét tạm ngừng điều trị Stivarga
Xuất huyết
Stivarga có liên quan đến tăng tần suất các biến cố chảy máu, đôi khi gây tử vong, (xem phần ‘Tác dụng không mong muốn’). Cần giám sát số lượng các tế bào máu và các thông số đông máu trên những bệnh nhân có yếu tố nguy cơ chảy máu, và trên những bệnh nhân điều trị với các chất chống đông (như warfarin) hoặc dùng các sản phẩm điều trị đồng thời khác làm tăng nguy cơ chảy máu. Việc sàng lọc cho và để điều trị tiếp những bệnh nhân giãn tĩnh mạch thực quản trên bệnh nhân xơ gan nên được thực hiện như một chăm sóc thường quy trước khi tiến hành điều trị với Stivarga. Trong các trường hợp chảy máu cần can thiệp y tế khẩn cấp, nên cân nhắc ngừng hẳn Stivarga
Thủng và rò đường tiêu hóa
Thủng đường tiêu hóa (bao gồm cả các trường hợp bị tử vong) đã được báo cáo ở những bệnh nhân được điều trị với Stivarga (xem phần ‘Tác dụng không mong muốn’). Những biến cố này cũng được biết là các biến chứng thường gặp liên quan đến bệnh ở những bệnh nhân có khối u ác tính trong ổ bụng. Cần ngừng Stivarga ở những bệnh nhân bị thủng hoặc có lỗ rò đường tiêu hóa. Chưa biết về tính an toàn của việc bắt đầu sử dụng lại Stivarga sau khi bị thủng hoặc rò đường tiêu hóa.
Thiếu máu cục bộ và nhồi máu cơ tim
Stivarga có liên quan với sự gia tăng tần suất mắc mới bệnh tim thiếu máu cục bộ và nhồi máu cơ tim (xem phần “Tác dụng không mong muốn”). Bệnh nhân đau thắt ngực không ổn định hoặc cơn đau thắt ngực mới khởi phát (trong vòng 3 tháng bắt đầu điều trị Stivarga), nhồi máu cơ tim gần đây (trong vòng 6 tháng bắt đầu điều trị Stivarga) và bệnh nhân có phân độ suy tim NYHA II hoặc cao hơn bị loại khỏi nghiên cứu lâm sàng
Bệnh nhân có tiền sử bệnh tim thiếu máu cục bộ cần được theo dõi các dấu hiệu và triệu chứng lâm sàng của bệnhnày. Ở những bệnh nhân có bệnh tim thiếu máu cục bộ và/hoặc nhồi máu cơ tim tiến triển, được khuyến cáo ngừng sử dụng Stivarga cho đến khi bệnh được giải quyết. Quyết định tái điều trị Stivarga phải dựa trên xem xét cẩn thận những lợi ích và rủi ro tiềm tàng của từng bệnh nhân. Nên chấm dứt điều trị Stivarga nếu không có hướng giải quyết.
Không quan sát thấy sự khác biệt giữa Stivarga và giả dược về tần suất loạn nhịp tim hoặc suy tim có ý nghĩa lâm sàng
Hội chứng bệnh chất trắng não sau có hồi phục (Reversible Posterior Leukoencephalopathy Syndrome)
Hội chứng bệnh chất trắng não sau có hồi phục (Reversible Posterior Leukoencephalopathy Syndrome - RPLS) đã được báo cáo có liên quan với việc điều trị Stivarga (xem phần “ Tác dụng không mong muốn”. Các dấu hiệu và triệu chứng của RPLS bao gồm co giật, đau đầu, tình trạng rối loạn tâm thần, rối loạn thị giác hoặc mù vỏ não, có hoặc không có kèm theo tăng huyết áp. Chẩn đoán xác định RPLS cần chụp ảnh não. Ở những bệnh nhân xuất hiện RPLS, ngừng sử dụng Stivarga, cùng với kiểm soát tăng huyết áp và điều trị hỗ trợ các triệu chứng khác. Chưa rõ tính an toàn của việc bắt đầu sử dụng lại liệu pháp Stivarga ở những bệnh nhân trước đó trải qua RPLS.
Tăng huyết áp
Stivarga có liên quan với tăng tần suất mắc tăng huyết áp (xem phần ‘Tác dụng không mong muốn’). Cần kiểm soát huyết áp trước khi bắt đầu điều trị với Stivarga. Khuyến cáo theo dõi huyết áp và điều trị tăng huyết áp theo hướng dẫn điều trị chuẩn. Trong trường hợp tăng huyết áp nặng hay kéo dài cho dù đã điều trị đầy đủ, nên tạm ngừng Stivarga và/hoặc giảm liều theo quyết định của bác sĩ điều trị (xem phần ‘Liều lượng và cách dùng’ – mục ‘Hiệu chỉnh liều’). Trong trường hợp của tăng huyết áp kịch phát, nên ngừng sử dụng Stivarga.
Các biến chứng trên việc làm lành vết thương
Do các sản phẩm điều trị có đặc tính chống tạo mạch có thể ức chế hoặc ảnh hưởng đến sự làm lành vết thương, khuyến cáo tạm thời ngừng Stivarga vì lý do thận trọng ở những bệnh nhân phải có phẫu thuật lớn. Quyết định tiếp tục điều trị Stivarga sau một can thiệp phẫu thuật lớn cần phải dựa trên đánh giá lâm sàng về tình trạng lành của vết thương.
Độc tính trên da
Phản ứng da bàn tay- bàn chân (HFSR/palmar-plantar erythrodysesthesia syndrome) và phát ban là các phản ứng có hại trên da thường gặp nhất của Stivarga (xem phần ‘Tác dụng không mong muốn’). Trong các nghiên cứu lâm sàng, tỉ lệ HFSR ghi nhận cao hơn trên người Châu Á (đặc biệt là người Nhật) so với chủng người da trắng khi điều trị với Stivarga (xem phần Liều lượng và cách sử dụng).
Các biện pháp phòng ngừa HFSR bao gồm điều trị các vết chai và sử dụng đệm giày và găng tay để ngăn chặn áp lực ấn mạnh lên gan bàn chân và lòng bàn tay. Điều trị HFSR có thể bao gồm việc sử dụng các loại kem có tác dụng tiêu sừng (ví dụ như kem có urê, salicylic acid, hoặc alpha hydroxyl acid- được bôi một cách vừa đủ chỉ trên các khu vực bị ảnh hưởng) và các loại kem dưỡng ẩm (được bôi rộng) để làm giảm triệu chứng. Giảm liều và/hoặc ngừng tạm thời Stivarga, hoặc trong các trường hợp nặng hay kéo dài, cần cân nhắc ngừng lâu dài Stivarga (xem phần ‘Liều lượng và cách dùng’ – mục ‘Hiệu chỉnh liều’).
Các bất thường về xét nghiệm sinh hóa và chuyển hóa
Stivarga có liên quan đến tăng tần suất có bất thường về điện giải (bao gồm hạ phosphate máu, hạ calci máu, hạ natri máu và hạ kali máu) và các bất thường về chuyển hóa (bao gồm cả tăng hoóc môn kích thích tuyến giáp, lipase và amylase). Những bất thường nói chung là từ nhẹ đến trung bình, không liên quan đến biểu hiện lâm sàng, và thường không cần tạm ngừng thuốc hoặc giảm liều. Khuyến cáo cần giám sát các thông số sinh hóa và chuyển hóa trong quá trình điều trị Stivarga và áp dụng các biện pháp điều trị thay thế phù hợp với các hướng dẫn thực hành lâm sàng nếu cần. Tạm ngừng hoặc giảm liều hoặc ngưng vĩnh viễn Stivarga cần được xem xét trong trường hợp bất thường đáng kể kéo dài hoặc tái phát (xem phần “Liều lượng và cách dùng " – mục ‘Hiệu chỉnh liều’).
Các thận trọng đối với bệnh lý cụ thể - ung thư biểu mô tế bào gan (HCC)
Trong nghiên cứu pha III then chốt, có đối chứng giả dược, bệnh nhân đã được điều trị trước đó với sorafenib.
Không có đủ dữ liệu về các bệnh nhân ngừng điều trị sorafenib do độc tính của sorafenib hoặc chỉ dung nạp sorafenib ở liều thấp (<400 mg mỗi ngày). Khả năng dung nạp của Stivarga ở những bệnh nhân này chưa được xác định.
Thông tin quan trọng về thành phần thuốc
Liều dùng hàng ngày 160 mg có chứa 2,427 mmol (hoặc 55,8mg) muối. Những bệnh nhân có chế độ ăn kiêng cần kiểm soát muối nên cân nhắc.
Chất ức chế /cảm ứng CYP3A4
Số liệu in vitro chỉ ra rằng regorafenib được chuyển hóa bởi cytochrome CYP3A4 và uridine diphosphate glucuronosyl transferase UGT1A9.
Dùng ketoconazole (400 mg trong 18 ngày), một chất ức chế CYP3A4 mạnh, với một liều duy nhất regorafenib (160 mg vào ngày 5) dẫn đến sự gia tăng phơi nhiễm regorafenib trung bình (AUC) khoảng 33%, và giảm sự phơi nhiễm của các chất chuyển hóa có hoạt tính, M-2 (N-oxit) và M-5 (N-oxide và N-desmethyl) trung bình khoảng 90%. Cần tránh sử dụng đồng thời thuốc ức chế mạnh hoạt tính CYP3A4 (ví dụ như clarithromycin, nước ép bưởi, itraconazole, ketoconazole, posaconazole, telithromycin và voriconazole) vì ảnh hưởng của chúng trên sự phơi nhiễm với trạng thái ổn định của regorafenib và các chất chuyển hóa của nó (M-2 và M -5) chưa được nghiên cứu.
Nên tránh dùng chung với các chất ức chế mạnh UGT1A9 (ví dụ mefenamic acid, diflunisal, and niflumic acid) trong quá trình điều trị với regorafenib vì ảnh hưởng của chúng lên sự phơi nhiễm ở trạng thái hằng định của regorafenib và các chất chuyển hóa của nó chưa được nghiên cứu. Dùng rifampin (600 mg trong 9 ngày), một chất cảm ứng CYP3A4 mạnh, với một liều duy nhất regorafenib (160 mg vào ngày 7) dẫn đến làm giảm AUC của regorafenib trung bình khoảng 50%, tăng 3 đến 4 lần phơi nhiễm trung bình của chất chuyển hóa có hoạt tính M-5, và không thay đổi phơi nhiễm của chất chuyển hóa có hoạt tính M-2. Các chất cảm ứng mạnh hoạt tính CYP3A4 khác (ví dụ như phenytoin, carbamazepine, phenobarbital) cũng có thể làm tăng chuyển hóa regorafenib. Do sự giảm nồng độ trong huyết tương của regorafenib có thể dẫn đến giảm hiệu quả, cần tránh sử dụng các chất gây cảm ứng mạnh CYP3A4, hoặc nên xem xét lựa chọn một thuốc thay thế khi dùng đồng thời, không có hoặc rất ít có khả năng cảm ứng CYP3A4.
Các chất ức chế/cảm ứng protein kháng ung thư vú (Breast cancer resistance protein - BCRP) và P-glycoprotein
Các dữ liệu in vitro chỉ ra rằng các chất chuyển hóa có hoạt tính M-2 và M-5 là cơ chất của BCRP và P-glycoprotein. Các chất ức chế và cảm ứng BRCP và P-glycoprotein có thể ảnh hưởng tới nồng độ của M-2 và M-5. Chưa rõ ý nghĩa lâm sàng của những phát hiện này.
Cơ chất UGT1A1 và UGT1A9
Số liệu in vitro cho thấy regorafenib cũng như các chất chuyển hóa hoạt động của nó M-2 ức chế glucoronin hóa trung gian bởi uridine transferases glucuronosyl diphosphate UGT1A1 và UGT1A9, trong khi M-5 chỉ ức chế UGT1A1 ở nồng độ đạt được trong cơ thể ở trạng thái ổn định.
Dùng regorafenib với một khoảng nghỉ dài 5 ngày trước khi dùng irinotecan làm tăng khoảng 44% AUC trung bình của SN-38, một cơ chất của UGT1A1 và là một chất chuyển hóa có hoạt tính của irinotecan. Cũng quan sát thấy có sự gia tăng phơi nhiễm trung bình với irinotecan khoảng 28%. Điều này cho thấy rằng việc sử dụng đồng thời regorafenib có thể làm tăng phơi nhiễm hệ thống với các cơ chất của UGT1A1 và UGT1A9.
Cơ chất BCRP (Breast cancer resistance protein : protein kháng ung thư vú) và P-glycoprotein
Dùng regorafenib (160 mg trong 14 ngày) trước khi dùng liều đơn rosuvastatin (5mg), là cơ chất của BCRP, làm tăng nồng độ AUC trung bình và Cmax của rosuvastatin tương ứng là 3,8 lần và 4,6 lần.
Điều này chỉ ra rằng dùng cùng regorafenib có thể làm tăng nồng độ huyết tương của các cơ chất của BCRP (ví dụ methotrexate, fluvastatin, atorvastatin). Vì vậy, nên theo dõi bệnh nhân một cách chặt chẽ về các dấu hiệu và triệu chứng của việc tăng nồng độ các cơ chất của BCRP
Dữ liệu lâm sàng chỉ ra rằng regorafenib không có tác động lên dược động học của digoxin, vì vậy regorafenib có thể được dùng cùng với các cơ chất p-glycoprotein, như digoxin, mà không có tương tác thuốc có ý nghĩa lâm sàng.
Cơ chất chọn lọc của các phân nhóm CYP
Dữ liệu in vitro cho thấy rằng regorafenib là một chất ức chế cạnh tranh của các cytochrome CYP2C8, CYP2C9, CYP2B6 ở nồng độ đạt được in vivo ở trạng thái ổn định (nồng độ đỉnh trong huyết tương là 8,1 micromolar). Tiềm năng ức chế in vitro đối với CYP3A4 và CYP2C19 ít rõ ràng hơn.
Một nghiên cứu lâm sàng thăm dò cơ chất đã được thực hiện để đánh giá hiệu quả của 14 ngày dùng regorafenib với liều 160 mg trên dược động học các cơ chất của CYP2C8 (rosiglitazone), CYP2C9 (S-warfarin), CYP2C19 (omeprazole) và CYP3A4 (midazolam). Dữ liệu dược động học cho thấy rằng regorafenib có thể được dùng đồng thời với các cơ chất CYP2C8, CYP2C9, CYP3A4, và CYP2C19 mà không có tương tác thuốc có ý nghĩa lâm sàng (xem thêm phần 'Cảnh báo đặc biệt và thận trọng').
Kháng sinh
Phân tích nồng độ - thời gian cho thấy regorafenib và các chất chuyển hóa của nó có thể trải qua tuần hoàn gan-ruột (xem phần “Các đặc tính dược động học”). Việc dùng cùng với neomycin, thuốc chống nhiễm khuẩn hấp thụ kém dùng trong nhiễm trùng đường ruột (có thể ảnh hưởng đến vòng tuần hoàn gan ruột của regorafenib) không ảnh hưởng đến nồng độ regorafenib. Có sự giảm đáng kể nồng độ của các chất chuyển hóa có hoạt tính M-2 và M-5. Tác động của các kháng sinh khác chưa được nghiên cứu. Ý nghĩa lâm sàng của tương tác với neomycin và những tương tác tiềm tàng với các kháng sinh khác là không rõ, nhưng có thể làm giảm hiệu quả của Stivarga.
Tương tác dược động học với các kháng sinh khác chưa được nghiên cứu.
Các thuốc gắn acid mật
Regorafenib , M-2 và M-5 có khả năng trải qua tuần hoàn gan-ruột (xem phần 5.2). Các thuốc gắn acid mật như cholestyramin và cholestagel có thể tương tác với regorafenib bằng cách hình thành phức hợp không hòa tan có thể ảnh hưởng đến sự hấp thu (hoặc tái hấp thu), vì vậy dẫn đến khả năng giảm sự phơi nhiễm. Ý nghĩa lâm sàng của những tương tác tiềm tàng này là không rõ, nhưng có thể dẫn đến giảm hiệu quả của regorafenib.
Phụ nữ có thai
Không có dữ liệu về việc sử dụng của regorafenib ở phụ nữ mang thai.
Dựa trên cơ chế tác dụng, regorafenib bị nghi ngờ gây ra tổn hại cho thai khi dùng trong thời gian mang thai.
Các nghiên cứu trên động vật đã cho thấy có độc tính trên sinh sản (xem dữ liệu ‘Dữ liệu an toàn tiền lâm sàng’).
Không nên sử dụng Stivarga trong thời kỳ mang thai, trừ khi rõ ràng là cần thiết và sau khi xem xét cẩn thận những lợi ích cho mẹ và nguy cơ cho thai nhi.
Cho con bú
Chưa biết liệu regorafenib/chất chuyển hóa có được bài tiết trong sữa mẹ hay không.
Ở chuột, regorafenib/chất chuyển hóa được bài tiết qua sữa.
Không thể loại trừ nguy cơ cho trẻ bú mẹ. Regorafenib có thể gây hại cho sự tăng trưởng và phát triển của trẻ em (xem dữ liệu ‘Dữ liệu an toàn tiền lâm sàng’). Phải ngừng cho con bú trong thời gian điều trị với Stivarga.
Khả năng sinh sản
Không có dữ liệu về tác dụng của Stivarga trên khả năng sinh sản ở người. Kết quả nghiên cứu trên động vật cho thấy regorafenib có thể làm giảm khả năng sinh sản của động vật đực và cái (xem phần ‘Dữ liệu an toàn tiền lâm sàng’).
Tránh thai
Phụ nữ có khả năng mang thai phải được thông báo rằng regorafenib có thể gây hại cho thai.
Phụ nữ có khả năng mang thai và nam giới nên đảm bảo sử dụng biện pháp tránh thai hiệu quả trong quá trình điều trị và kéo dài đến 8 tuần sau khi hoàn tất điều trị.
Không có nghiên cứu về những ảnh hưởng của Stivarga trên khả năng lái xe hoặc sử dụng máy móc. Nếu bệnh nhân có các triệu chứng ảnh hưởng đến khả năng tập trung và phản ứng khi điều trị với Stivarga, họ không được lái xe hay vận hành máy móc cho đến khi các ảnh hưởng giảm bớt.
Tóm tắt thông tin về an toàn
Thông tin về an toàn tổng thể của Stivarga được dựa trên dữ liệu từ hơn 4.800 bệnh nhân được điều trị trong các thử nghiệm lâm sàng bao gồm dữ liệu pha III có đối chứng giả dược trên 636 bệnh nhân bị ung thư đại trực tràng di căn (CRC), 132 bệnh nhân có u mô đệm đường tiêu hóa (GIST) và 374 bệnh nhân bị ung thư tế bào gan (HCC).
Dữ liệu an toàn của regorafenib trong những nghiên cứu này phù hợp với kết quả về tính an toàn của nghiên cứu pha III B tiến hành trên 2872 bệnh nhân bị ung thư đại trực tràng di căn có bệnh tiến triển sau khi điều trị với các liệu pháp chuẩn.
Các phản ứng bất lợi nghiêm trọng nhất ở những bệnh nhân dùng Stivarga là tổn thương gan nặng, xuất huyết, thủng đường tiêu hóa và nhiễm trùng.
Phản ứng bất lợi thường gặp nhất (≥ 30%) ở bệnh nhân dùng Stivarga là đau, phản ứng da chân-tay, suy nhược/ mệt mỏi, tiêu chảy, giảm sự thèm ăn và giảm lượng thức ăn, tăng huyết áp, và nhiễm trùng.
Bảng các phản ứng bất lợi
Các phản ứng bất lợi được báo cáo trong các nghiên cứu lâm sàng trên các bệnh nhân điều trị bằng Stivarga được thể hiện trong Bảng 3. Chúng được phân loại theo các hệ cơ quan. Thuật ngữ MedDRA thích hợp nhất được sử dụng để mô tả một phản ứng nhất định và các từ đồng nghĩa của nó và bệnh lý liên quan.
Phản ứng bất lợi của thuốc được phân nhóm theo tần suất. Các nhóm tần suất được xác định bởi các quy ước sau đây: rất thường gặp: ≥ 1/10; thường gặp: 1/100 đến <1/10; ít gặp: ≥ 1/1, 000 đến <1/100; hiếm gặp: 1/10, 000 đến <1 / 1.000.
Trong mỗi nhóm tần suất, các tác dụng không mong muốn được trình bày theo thứ tự giảm dần về mức độ nghiêm trọng.
Bảng 3: Các phản ứng bất lợi được báo cáo trong các thử nghiệm lâm sàng trên các bệnh nhân điều trị với Stivarga
|
Các hệ cơ quan |
Rất thường gặp |
Thường gặp |
Ít gặp |
Hiếm gặp |
|
Nhiễm trùng và nhiễm ký sinh trùng |
Nhiễm trùng* |
|
|
|
|
Bướu lành, ác tính và không đặc hiệu (bao gồm nang và polyp) |
|
|
|
U gai sừng/ Ung thư biểu mô tế bào vảy của da |
|
Các rối loạn máu và hệ bạch huyết |
Giảm tiểu cầu Thiếu máu |
Giảm bạch cầu |
|
|
|
Các rối loạn hệ miễn dịch |
|
|
Phản ứng quá mẫn |
|
|
Các rối loạn hệ nội tiết |
|
Suy giáp |
|
|
|
Các rối loạn dinh dưỡng và chuyển hóa |
Giảm thèm ăn và lượng thức ăn |
Hạ kali máu Hạ phosphat máu hạ calci máu Hạ natri máu Hạ magnesi máu Tăng acid uric máu |
|
|
|
Các rối loạn hệ thần kinh |
|
Đau đầu Run |
|
Hội chứng bệnh chất trắng não sau có hồi phục (RPLS) |
|
Các rối loạn ở tim |
|
|
Nhồi máu cơ tim Thiếu máu cục bộ cơ tim |
|
|
Các rối loạn ở mạch |
Xuất huyết* Tăng huyết áp |
|
Tăng huyết áp kịch phát |
|
|
Các rối loạn hô hấp, ở lồng ngực và trung thất |
Mất giọng |
|
|
|
|
Các rối loạn hệ tiêu hóa |
Tiêu chảy Viêm miệng Nôn Buồn nôn
|
Rối loạn vị giác Khô miệng Trào ngược dạ dày – thực quản Viêm dạ dày ruột |
Thủng đường tiêu hóa* Rò đường tiêu hóa Viêm tụy
|
|
|
Các rối loạn gan mật |
Tăng bilirubin máu Tăng các transaminase |
|
Tổn thương gan nặng *# |
|
|
Các rối loạn da và mô dưới da |
Phản ứng da chân- tay** Ban đỏ
|
Rụng tóc Khô da Ban tróc da |
Rối loạn móng Hồng ban đa dạng |
Hội chứng Stevens-Johnson Hoại tử thượng bì nhiễm độc |
|
Các rối loạn hệ cơ xương và mô liên kết |
|
Cứng cơ xương |
|
|
|
Các rối loạn thận và tiết niệu |
|
Protein niệu |
|
|
|
Các rối loạn toàn thân và tại nơi dùng thuốc |
Suy nhược/ Đau Sốt Viêm niêm mạc |
|
|
|
|
Các xét nghiệm |
Giảm cân |
Tăng amylase Tăng lipase INR bất thường |
|
|
*Một số ca tử vong đã được báo cáo
** hội chứng palmar-plantar erythrodysesthesia trong thuật ngữ MedDRA
# theo tiêu chuẩn tổn thương gan do thuốc (drug-induced liver injury -DILI) của nhóm chuyên gia DILI quốc tế
Mô tả một số phản ứng bất lợi
Xuất huyết
Trong các thử nghiệm pha III có đối chứng giả dược, tỷ lệ chung của biến cố xuất huyết/ chảy máu là 18,2% ở những bệnh nhân được điều trị với Stivarga và 9,5% ở những bệnh nhân điều trị với giả dược. Hầu hết các trường hợp có biến cố chảy máu ở bệnh nhân điều trị bằng Stivarga có mức độ từ nhẹ đến trung bình (độ 1 và 2: 15,2%), đáng chú ý nhất là chảy máu cam (6,1%). Biến cố gây tử vong trên các bệnh nhân được điều trị bằng Stivarga ít gặp (0,7%), và bao gồm biến cố ở não, ở đường hô hấp, tiêu hóa và sinh dục.
Nhiễm trùng
Trong các thử nghiệm pha III có đối chứng giả dược, bệnh nhân điều trị với Stivarga bị nhiễm trùng nhiều hơn so với bệnh nhân dùng giả dược (tất cả các độ: 31,6% so với 17,2%). Hầu hết các nhiễm trùng ở những bệnh nhân được điều trị với Stivarga có mức độ từ nhẹ đến trung bình (độ 1 và 2: 22,9%), và bao gồm nhiễm trùng đường tiểu (5,7%), viêm vòm họng (4,0%), nhiễm nấm ở niêm mạc và nhiễm nấm toàn thân (3,3%) và viêm phổi (2,6%). Biến cố tử vong do nhiễm trùng được ghi nhận nhiều hơn trên những bệnh nhân điều trị với Stivarga (1,0%) so với những bệnh nhân dùng giả dược (0,3%) và phần lớn là các biến cố đường hô hấp.
Phản ứng da bàn tay – chân
Trong các thử nghiệm pha III có đối chứng giả dược, phản ứng trên da ở bàn tay bàn chân (HFSR) được quan sát thấy thường gặp hơn ở những bệnh nhân được điều trị bằng Stivarga so với bệnh nhân dùng giả dược (tất cả các độ: 51,4%, so với 6,5% trên bệnh nhân CRC, 66.7% so với 15.2 % trên bệnh nhân GIST và 51,3% so với 7,3% trên bệnh nhân HCC). Hầu hết các trường hợp HFSR ở những bệnh nhân được điều trị với Stivarga xuất hiện trong chu kỳ điều trị đầu tiên và có mức độ từ nhẹ đến trung bình (Độ 1 và 2: 34,3% CRC, 44,7% GIST và 39,3% HCC). Tỷ lệ HFSR Độ 3 là 17,1% (CRC), 22% (GIST) và 12,3% (HCC). Tỷ lệ bị HFSR ở bệnh nhân Châu Á khi dùng Stivarga là cao hơn (tất cả các độ: 74,8% CRC, 88.2% GIST và 67,1% HCC và Độ 3: 20,5% CRC, 23.5%, GIST và 13,5% HCC) (xem mục 'Liều dùng và cách dùng '- tiểu mục " Thông tin bổ sung về các đối tượng bệnh nhân đặc biệt')
Tăng huyết áp
Trong các thử nghiệm pha III có đối chứng giả dược, tỷ lệ chung của tăng huyết áp cao hơn ở những bệnh nhân điều trị với Stivarga so với những bệnh nhân dùng giả dược (29,6% so với 7,5% CRC, 60,6% so với 25,8% GIST và 31% so với 6,2% HCC). Hầu hết các trường hợp tăng huyết áp ở bệnh nhân được điều trị với Stivarga xuất hiện trong chu kỳ điều trị đầu tiên và có mức độ từ nhẹ đến trung bình (độ 1 và 2: 20,9% CRC, 31,8% GIST và 15,8% HCC). Tỷ lệ tăng huyết áp độ 3 là 8,7%% (CRC) và 28,0% (GIST) và 15,2% (HCC). Một trường hợp tăng huyết áp độ 4 được báo cáo trong thử nghiệm trên bệnh nhân GIST.
Tổn thương gan nặng
Trong phần lớn các trường hợp tổn thương gan nặng, có khởi phát rối loạn chức năng gan trong vòng 2 tháng điều trị đầu tiên, và có đặc trưng là kiểu tổn thương tế bào gan có tăng các men transaminase > 20 lần giới hạn trên bình thường, tiếp theo đó là tăng bilirubin. Trong các thử nghiệm lâm sàng, đã quan sát thấy tổn thương gan nặng gây tử vong với một tỷ lệ cao hơn ở những bệnh nhân điều trị bằng Stivarga người Nhật Bản (~ 1,5%) so với bệnh nhân không phải người Nhật bản (<0,1%).
Trong các nghiên cứu lâm sàng pha III có đối chứng giả dược, tỉ lệ chung protein niệu cần điều trị khẩn là 9,1% ở những bệnh nhân điều trị với Stivarga, so với 1,9% những bệnh nhân điều trị với giả dược. Ở những trường hợp này, 35,6% ở nhóm dùng Stivarga và 54,5% ở nhóm dùng giả dược được ghi nhận là không hồi phục.
Trong tất cả các nghiên cứu lâm sàng, các biến cố rối loạn tim mạch (mọi cấp độ) được ghi nhận thường xuyên hơn (13,7% so với 6,5%) ở những bệnh nhân điều trị với Stivarga có tuổi từ 75 trở lên (N=410) so với những bệnh nhân điều trị với Stivarga tuổi dưới 75 (N=4108).
Các bất thường về xét nghiệm
Những bất thường về xét nghiệm liên quan đến điều trị được quan sát thấy trong các thử nghiệm pha III có đối chứng giả dược được trình bày trong Bảng 4, bảng 4a, (xem thêm phần 'Cảnh báo đặc biệt và thận trọng'’).
Bảng 4: Những bất thường cần điều trị khẩn ghi nhận trong các nghiên cứu lâm sàng pha III có đối chứng giả dược trên những bệnh nhân CRC di căn (CORRECT), GIST (GRID) và HCC (RESORCE)
|
|
mCRC (CORRECT) |
GIST (GRID) |
HCC (RESORCE) |
|||||||||
|
Các thông số xét nghiệm |
Stivarga phối hợp |
Giả dược phối hợp BSC (n=253) |
Stivarga phối hợp BSC (n= 500) |
Giả dược phối hợp BSC (n=253) |
Stivarga phối hợp |
Giả dược phối hợp BSC (n= 66) |
Stivarga phối hợp BSC (n=132) |
Giả dược phối hợp BSC (n= 66) |
Stivarga phối hợp BSC |
Giả dược phối hợp BSC (n=193) |
Stivarga phối hợp BSC (n= 374) |
Giả dược phối hợp BSC (n=193) |
|
Độ a |
Độ b |
Độ b |
||||||||||
|
Tất cả các mức độ % |
Độ 3/4 % |
Tất cả các mức độ % |
Độ 3/4 % |
Tất cả các mức độ % |
Độ 3/4 % |
|||||||
|
Các rối loạn máu và hệ bạch huyết Giảm hemoglobin Giảm tiểu cầu Giảm bạch cầu trung tính Giảm bạch cầu lympho |
78.5
40.5 2.8 54.1 |
66.3
16.8 0 34.8 |
5.3
2.8 0.6 9.3 |
2.8
0.4 0 4.0 |
75.0
12.9 15.9 29.9 |
72.7
1.5 12.1 24.2 |
3.0
0.8 3.1 7.6 |
1.5
1.5 3.0 3.0 |
72.5
63.1 13.6 67.8 |
71.3
50.0 14.9 58.5 |
6.0
5.4 3.0 17.4 |
4.8
0 1.0 11.7 |
|
Các rối loạn chuyển hóa và dinh dưỡng Giảm calcium Giảm kali Giảm phosphate |
59.3 25.7 57.4 |
18.3 8.3 11.1 |
1.2 4.3 31.1 |
1.2 0.4 3.6 |
16.7 20.5 54.5 |
4.5 3.0 3.1 |
1.5 3.0 21.2 |
0 0 1.5 |
23.4 30.7 70.4 |
10.1 9.0 31.4 |
0.3 4.3 33.9 |
0 2.1 6.9 |
|
Các rối loạn gan mật Tăng bilirubin Tăng AST Tăng ALT |
44.6 65.0 45.2 |
17.1 45.6 29.8 |
12.2 5.9 5.5 |
8.4 5.2 3.2 |
33.3 58.3 39.4 |
12.1 47.0 39.4 |
3.8 3.8 4.6 |
1.5 3.0 1.5 |
78.2 92.7 70.4 |
54.5 84.3 58.6 |
15.9 17.8 6.2 |
15.7 19.9 4.7 |
|
Các rối loạn thận và tiết niệu Protein niệu |
83.6 |
61.0 |
1.8 |
0.8 |
59.2 |
52.5 |
3.1 |
3.4 |
50.8 |
36.7 |
16.7 |
3.2 |
|
Các xét nghiệm thăm dò Tăng INR** Tăng lipase Tăng amylase |
23.7 46.0 25.5 |
16.6 18.7 16.7 |
4.2 11.4 2.6 |
1.6 4.4 2.4 |
9.3 14.4 - |
12.5 4.6 - |
1.6 0.8 - |
4.7 0 - |
44.2 40.5 23.0 |
35.4 27.0 19.0 |
0.7 14.2 2.8 |
2.1 8.7 2.7 |
a Tiêu chuẩn thuật ngữ chung về biến cố có hại (CTCAE). Phiên bản 3.0
b Tiêu chuẩn thuật ngữ chung về biến cố có hại (CTCAE), Phiên bản 4.0
* Chỉ số bình thường hóa quốc tế
BSC = Best Supportive Care: Chăm sóc hỗ trợ tốt nhất
So với thử nghiệm pha III toàn cầu trên bệnh nhân CRC (CORRECT) với chủ yếu (~ 80%) bệnh nhân người da trắng được thu nhận, đã quan sát thấy tăng enzym gan với một tỷ lệ cao hơn ở các bệnh nhân điều trị Stivarga trong thử nghiệm pha III CRC ở châu Á (CONCUR) với phần lớn (> 90 %) bệnh nhân ở Đông Á được thu nhận
Bảng 4a: Các trường hợp bất thường enzym gan có liên quan đến điều trị được báo cáo trong thử nghiệm pha III đối chứng giả dược ở các bệnh nhân Châu Á bị CRC di căn (CONCUR)
|
Thông số xét nghiệm, |
Stivarga phối hợp với BSC§ (N=136) |
Giả dược phối hợp với BSC§ (N=68) |
||||
|
Tất cả các độ*
|
Độ 3* |
Độ 4* |
Tất cả các độ*
|
Độ 3* |
Độ 4* |
|
|
Tăng bilirubin |
66.7 |
7.4 |
4.4 |
32.8 |
4.5 |
0.0 |
|
Tăng AST |
69.6 |
10.4 |
0.7 |
47.8 |
3.0 |
0.0 |
|
Tăng ALT |
54.1 |
8.9 |
0.0 |
29.9 |
1.5 |
0.0 |
§ Chăm sóc hỗ trợ tốt nhất (Best Supportive Care)
* Tiêu chuẩn thuật ngữ chung về biến cố bất lợi , phiên bản 4.0
Trong các thử nghiệm lâm sàng pha III có đối chứng giả dược, các xét nghiệm hormone kích thích tuyến giáp (TSH) cho thấy giá trị so với giá trị ban đầu > ULN (giá trị bình thường giới hạn trên) ở 34,6% bệnh nhân điều trị với Stivarga và 17,2% bệnh nhân điều trị giả dược. Chỉ số TSH so với giá trị ban đầu > 4 lần ULN đã được ghi nhận ở 6,5% bệnh nhân điều trị với Stivarga và ở 1,3% bệnh nhân điều trị giả dược. Nồng độ triiodothyronine (FT3) tự do so với thời điểm ban đầu thấp hơn giới hạn dưới của giá trị bình thường (<LLN) được ghi nhận ở 29,2% bệnh nhân điều trị với Stivarga và ở 20,4% bệnh nhân điều trị với giả dược. Nồng độ thyroxin tự do (FT4) so với giá trị ban đầu < LLN được ghi nhận ở 8,1% bệnh nhân điều trị với Stivarga và 5,6% bệnh nhân điều trị với giả dược. Nhìn chung có khoảng 4,6% bệnh nhân điều trị với Stivarga bị suy tuyến giáp cần điều trị hormone thay thế.
Báo cáo các trường hợp nghi ngờ phản ứng bất lợi
Việc báo cáo các trường hợp nghi ngờ phản ứng bất lợi sau khi thuốc lưu hành là quan trọng. Nó cho phép theo dõi liên tục cán cân lợi ích/ nguy cơ của sản phẩm. Các bác sĩ cần báo cáo tất cả các trường hợp nghi ngờ phản ứng bất lợi.
Liều cao nhất của Stivarga được nghiên cứu trên lâm sàng là 220 mg mỗi ngày. Phản ứng bất lợi của thuốc thường gặp nhất ở liều này là các biến cố trên da, mất giọng, tiêu chảy, viêm niêm mạc, khô miệng, chán ăn, tăng huyết áp, và mệt mỏi.
Không có thuốc giải độc đặc hiệu để điều trị quá liều Stivarga. Trong trường hợp nghi ngờ quá liều, nên ngừng Stivarga ngay lập tức, với chăm sóc hỗ trợ tốt nhất được áp dụng bởi một chuyên gia y tế, và bệnh nhân cần được theo dõi cho đến khi ổn định lâm sàng.
Các đặc tính dược lực học:
Nhóm dược lý: các tác nhân chống ung thư, chất ức chế protein kinase
Mã ATC: L01XE21
Cơ chế tác động và tác dụng dược lực học
Regorafenib là một tác nhân bất hoạt khối u đường uống, ức chế mạnh nhiều protein kinase, bao gồm cả các kinase liên quan đến sự tân sinh mạch ở khối u (VEGFR1, -2, -3, TIE2), sinh ung thư (KIT, RET, RAF-1, BRAF, BRAFV600E) , di căn (VEGFR3, PDGFR, FGFR) và miễn dịch khối u (CSF1R). Đặc biệt, regorafenib ức chế đột biến KIT, một gen gây ung thư chủ yếu trong u mô đệm đường tiêu hóa, và do đó ức chế tăng sinh tế bào khối u. Trong các nghiên cứu tiền lâm sàng, regorafenib đã được chứng minh có hoạt tính chống ung thư mạnh trên một số lượng lớn các mô hình khối u bao gồm các khối u đại trực tràng và u mô đệm đường tiêu hóa và u tế bào biểu mô gan được thể hiện dưới các tác dụng chống tạo mạch và chống tăng sinh. Ngoài ra, regorafenib làm giảm nồng độ đại thực bào liên quan khối u và cho thấy tác dụng chống di căn in vivo. Chất chuyển hóa chính ở người (M-2 và M-5) thể hiện hiệu quả tương tự so với regorafenib in vitro và trong các mô hình in vivo.
Hiệu quả và an toàn trên lâm sàng
Ung thư đại trực tràng (Colorectal cancer -CRC) di căn
Hiệu quả lâm sàng và an toàn của Stivarga đã được đánh giá trong một nghiên cứu pha III quốc tế, đa trung tâm, ngẫu nhiên, mù đôi, có đối chứng giả dược (CORRECT) ở những bệnh nhân đã được điều trị tích cực trước đó, bị ung thư đại trực tràng di căn đã tiến triển sau khi thất bại với điều trị chuẩn.
Tiêu chí hiệu quả chính là thời gian sống còn toàn bộ (Overall Survival - OS). Tiêu chí phụ là thời gian sống còn không bệnh tiến triển (Progression-Free Survival -PFS), tỷ lệ đáp ứng khối u (Objective tumor Response Rate - ORR) và tỷ lệ kiểm soát bệnh (Disease Control Rate DCR).
Tổng cộng, 760 bệnh nhân được phân ngẫu nhiên theo tỷ lệ 2:1 để nhận 160 mg regorafenib (4 viên Stivarga mỗi viên có chứa 40 mg regorafenib) đường uống một lần mỗi ngày (N = 505) phối hợp với chăm sóc hỗ trợ tốt nhất (Best Supportive Care -BSC) hoặc giả dược tương ứng (N = 255) phối hợp với BSC trong 3 tuần sau đó có 1 tuần ngừng điều trị. Liều trung bình hàng ngày regorafenib là 147 mg.
Bệnh nhân tiếp tục điều trị cho đến khi bệnh tiến triển hoặc có độc tính không thể chấp nhận được. Một phân tích tạm thời đã được lên kế hoạch trước để đánh giá về hiệu quả, được thực hiện khi 432 trường hợp tử vong xảy ra. Nghiên cứu được mở mù sau khi phân tích tạm thời về OS này đã vượt qua ngưỡng hiệu quả xác định trước.
Trong số 760 bệnh nhân được phân ngẫu nhiên, giá trị trung vị của tuổi là 61 năm, 61% là nam giới, 78% là người da trắng, và tất cả các bệnh nhân có chỉ số toàn trạng ECOG (Performance Status -PS) trước điều trị là 0 hoặc 1. Các bệnh nhân PS≥2 được ghi nhận ở 11,4% bệnh nhân trong quá trình điều trị Stivarga. Thời gian điều trị trung bình và liều hàng ngày, cũng như tỉ lệ thay đổi liều và giảm liều trên những bệnh nhân được ghi nhận có PS≥2 điều trị với giả dược là 8,3%. Phần lớn bệnh nhân có PS≥2 ngừng điều trị vì bệnh tiến triển. Các vị trí khối u chính là đại tràng (65%), trực tràng (29%), hoặc cả hai (6%). 57% bệnh nhân có đột biến KRAS lúc khởi đầu nghiên cứu.
Phần lớn các bệnh nhân (52%) đã dùng 3 hoặc ít hơn các phác đồ điều trị bệnh di căn trước đó. Phương pháp điều trị bao gồm điều trị bằng hóa trị liệu có fluoropyrimidine, liệu pháp kháng VEGF, và, nếu bệnh nhân có loại KRAS hoang dã, một liệu pháp kháng EGFR.
Việc đưa thêm Stivarga vào BSC làm kéo dài thời gian sống thêm đáng kể hơn so với giả dược cộng với BSC với giá trị p= 0.005178 log rank test phân tầng, tỷ số nguy cơ (hazard ratio) là 0,774 [95% CI 0.636, 0.942] và giá trị trung vị của OS là 6,4 tháng so với 5,0 tháng (xem Bảng 5 và Hình 1). PFS dài hơn đáng kể ở những bệnh nhân dùng Stivarga cộng với BSC (hazard ratio: 0,494, p <0,000001, xem Bảng 5 và Hình 2). Tỉ lệ đáp ứng (đáp ứng một phần và đáp ứng toàn bộ) là 1% và 0,4% tương ứng với Stivarga và giả dược (p=0.188432, 1 phía ). Tỉ lệ kiểm soát bệnh (đáp ứng toàn bộ hoặc đáp ứng một phần hoặc bệnh ổn định) tăng đáng kể ở bệnh nhân điều trị với Stivarga (41,0% so với 14,9%, p < p<0.000001, 1 phía).
Bảng 5: Kết quả về hiệu quả từ nghiên cứu CORRECT
|
Các thông số về hiệu quả |
Tỉ số nguy cơ* (95% CI) |
Giá trị P (một phía) |
Trung vị (95% CI) |
|
|
Stivarga và BSC (N=505) |
Giả dược và BSC (N=255) |
|||
|
Thời gian sống còn toàn bộ
|
0.774 (0.636, 0.942) |
0.005178 |
6.4 tháng (5.9, 7.3) |
5.0 tháng (4.4, 5.8) |
|
Thời gian sống còn không bệnh tiến triển ** |
0.494 (0.419, 0.582) |
<0.000001 |
1.9 tháng (1.9, 2.1) |
1.7 tháng (1.7, 1.7) |
§ Best supportive care (BSC): Chăm sóc hỗ trợ tốt nhất
* Hazard ratio (tỉ số nguy cơ) < 1 ưu thế cho Stivarga
** Dựa trên đánh giá của nghiên cứu viên về đáp ứng khối u
Hình 1: Đường cong Kaplan-Meier về thời gian sống còn toàn bộ (Overall Survival)

Các phân tích dưới nhóm về tỉ lệ sống còn toàn bộ và sống còn không bệnh tiến triển nói chung chia theo tuổi (<65; ≥65), giới tính, ECOG PS, các vị trí khối u chính, thời gian từ lúc chẩn đoán di căn đầu tiên, thời gian điều trị ung thư trước đó, và tình trạng kháng KRAS cho thấy hiệu quả điều trị nhiều hơn ở nhóm regorafenib so với nhóm giả dược.
Kết quả phân tích dưới nhóm dựa vào bệnh sử tình trạng đột biến KRAS cho thấy hiệu quả điều trị về tỉ lệ sống còn toàn bộ của regorafenib là hơn giả dược trên những bệnh nhân có khối u loại KRAS hoãng dã trong khi hiệu quả thấp hơn một cách đáng kể được ghi nhận ở những bệnh nhân có khối u đột biến KRAS; hiệu quả điều trị của PFS (sống không bệnh tiến triển) cũng được ghi nhận ủng hộ cho regorafenib không kể đến tình trạng đột biến KRAS. Tỉ số nguy cơ (CI 95%) của tỉ lệ sống còn toàn bộ là 0,653 (0,476 đến 0,895) cho những bệnh nhân có khối u loại KRAS hoang dã và 0,867 (0,670 đến 1,123) cho những bệnh nhân có khối u đột biến KRAS, không có bằng chứng về việc không đồng nhất trong hiệu quả điều trị (thử nghiệm tương tác không có ý nghĩa). Tỉ lệ nguy cơ (CI 95%) của tỉ lệ sống còn không bệnh tiến triển là 0,475 (0,362 đến 0,623) trên những bệnh nhân có khối u loại KRAS hoang dã và 0,525 (0,425 đến 0,649) trên những bệnh nhân có khối u đột biến KRAS.
Một nghiên cứu pha III khác, quốc tế, đa trung tâm, ngẫu nhiên, mù đôi, có đối chứng giả dược (CONCUR), đánh giá hiệu quả và an toàn của Stivarga trên 204 bệnh nhân châu Á (> 90% người Đông Á) bị ung thư đại trực tràng di căn đã được điều trị trước đó, những người đã bệnh tiến triển sau khi thất bại với hóa trị liệu với phác đồ có fluoropyrimidine. 122 bệnh nhân trong nghiên cứu CONCUR cũng đã được điều trị trước đó với các thuốc nhắm đích VEGF hoặc EGFR.
Tiêu chí hiệu quả chính là thời gian sống còn toàn bộ (OS). Việc thêm Stivarga vào phác đồ BSC làm kéo dài thời gian sống còn toàn bộ đáng kể, so với nhóm dùng giả dược cộng với BSC với một tỷ số nguy cơ là 0.550 (p = 0.000159 trong kiểm định log rank test phân tầng) và giá trị trung vị của OS là 8.8 tháng so với 6.3 tháng [95% CI 0.395, 0.765]. PFS cũng dài hơn đáng kể ở những bệnh nhân dùng Stivarga cộng BSC (tỷ số nguy cơ: 0.311, p <0.000001).
Tính an toàn của Stivarga so với giả dược trong nghiên cứu CONCUR là phù hợp với hồ sơ an toàn đã thấy trong nghiên cứu CORRECT.
Các khối u mô đệm đường tiêu hóa (GIST)
Hiệu quả và an toàn trên lâm sàng của Stivarga đã được đánh giá trong một nghiên cứu pha III quốc tế, đa trung tâm, ngẫu nhiên, mù đôi, có đối chứng giả dược ở những bệnh nhân có khối u mô đệm đường tiêu hóa (GIST) được điều trị trước đó với 2 thuốc ức chế tyrosine kinase (imatinib và sunitinib).
Việc phân tích tiêu chí hiệu quả chính Thời gian sống còn không bệnh tiến triển (Progression Free Survival -PFS) đã được tiến hành sau khi có 144 biến cố PFS (đánh giá mù tập trung). Các tiêu chí phụ bao gồm Thời gian đến khi tiến triển bệnh (Time To Progression - TTP) và Thời gian sống còn toàn bộ (Overall Survival - OS) (phân tích tạm thời) cũng được đánh giá.
Tổng cộng, 199 bệnh nhân bị GIST được phân ngẫu nhiên với tỷ lệ 2:1 để dùng hoặc 160 mg regorafenib đường uống mỗi ngày một lần phối hợp Chăm sóc hỗ trợ tốt nhất (Best Supportive Care -BSC; n = 133) hoặc giả dược phối hợp BSC (n = 66) trong vòng 3 tuần điều trị, tiếp theo là 1 tuần nghỉ điều trị. Liều regorafenib hàng ngày trung bình nhận được là 140 mg.
Bệnh nhân tiếp tục điều trị cho đến khi bệnh tiến triển hoặc có độc tính không thể chấp nhận được. Bệnh nhân dùng giả dược khi đã có bệnh tiến triển, được gợi ý dùng regorafenib nhãn mở (lựa chọn điều trị chéo). Bệnh nhân dùng regorafenib bị bệnh tiến triển và những bệnh nhân mà theo ý kiến của nhà nghiên cứu, việc điều trị với regorafenib có lợi ích lâm sàng, có cơ hội để tiếp tục dùng regorafenib nhãn mở.
Với 199 bệnh nhân được phân ngẫu nhiên, tuổi trung bình là 58 năm, 64% là nam giới, 68% là người da trắng, và tất cả các bệnh nhân có chỉ số toàn trạng (Performance Status) ECOG 0 hoặc 1 trước điều trị. Trung vị thời gian từ khi có bệnh tiến triển gần đây hoặc tái phát bệnh đến lúc phân nhóm ngẫu nhiên là 6 tuần.
Regorafenib phối hợp BSC làm kéo dài hơn đáng kể PFS so với giả dược phối hợp BSC với một tỷ số nguy cơ là 0.268 [khoảng tin cậy 95% 0.185, 0.388] và có trung vị PFS là 4.8 tháng so với 0.9 tháng (p <0.000001). Nguy cơ tương đối của bệnh tiến triển hoặc tử vong đã giảm khoảng 73.2% ở những bệnh nhân được điều trị regorafenib so với bệnh nhân dùng giả dược (xem Bảng 6, Hình 2). Sự gia tăng PFS không phụ thuộc tuổi tác, giới tính, vùng địa lý, nhóm thuốc đã điều trị trước đó và chỉ số toàn trạng ECOG.
TTP đã dài hơn đáng kể ở những bệnh nhân dùng regorafenib phối hợp BSC so với bệnh nhân dùng giả dược phối hợp với BSC với tỷ số nguy cơ là 0.248 [95% CI 0.170, 0.364], và giá trị trung vị của TTP là 5.4 tháng so với 0.9 tháng (p <0.000001) (xem Bảng 6).
Tỷ số nguy cơ của phân tích OS cho thấy xu hướng có hiệu quả điều trị tích cực (HR = 0.772 [95% CI, 0.423, 1.408]; p = 0.199; giá trị trung vị của OS không xác định được ở hai nhóm) cho dù có điều trị chéo sau tiến triển bệnh của 85% bệnh nhân ban đầu được phân ngẫu nhiên vào nhóm giả dược (xem Bảng 6, Hình 3).
Bảng 6: Các kết quả về hiệu quả từ nghiên cứu GRID
|
|
Tỷ số nguy cơ* (95% CI) |
giá trị P (một phía) |
Giá trị trung vị (khoảng tin cậy 95%) |
|
|
Stivarga phối hợp BSC§ (N=133) |
Giả dược phối hợp BSC§ (N=66) |
|||
|
Thời gian sống còn không bệnh tiến triển |
0.268 (0.185, 0.388) |
<0.000001 |
4.8 tháng (4.0, 5.7) |
0.9 tháng (0.9, 1.1) |
|
Thời gian đến khi bệnh tiến triển |
0.248 (0.170,0.364) |
<0.000001 |
5.4 tháng (4.1, 5.7) |
0.9 tháng (0.9, 1.1) |
|
Thời gian sống còn toàn bộ |
0.772 (0.423, 1.408) |
0.199 |
NR** |
NR** |
§ Chăm sóc hỗ trợ tốt nhất (Best Supportive Care –BSC)
* Tỷ số nguy cơ Hazard ratio < 1 tốt hơn cho Stivarga
** NR: không xác định được
Hình 2: Đường cong Kaplan Meier của Thời gian sống còn không bệnh tiến triển
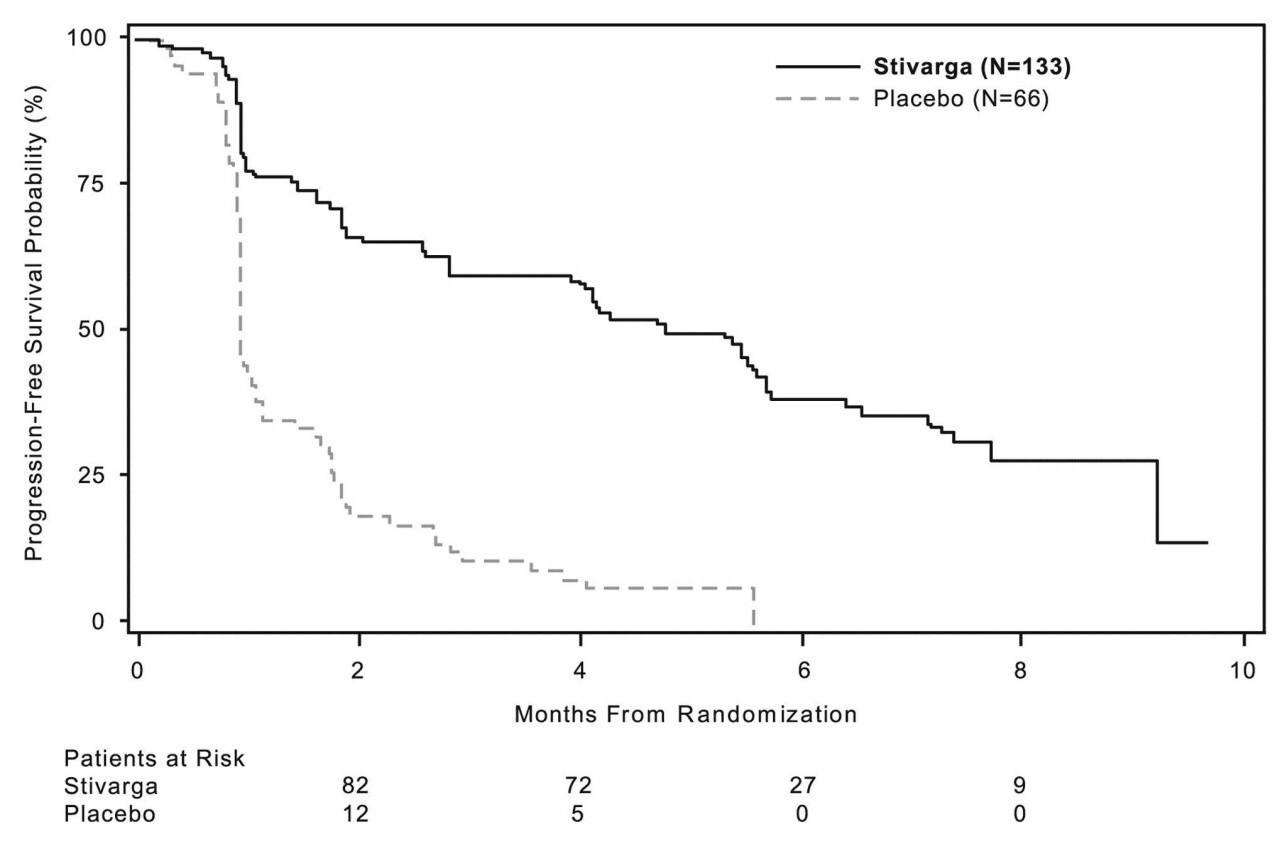
Hình 3: Đường cong Kaplan Meier của Thời gian sống còn toàn bộ

Ngoài ra, 56 bệnh nhân ở nhóm giả dược phối hợp với BSC được dùng Stivarga nhãn mở sau khi đổi chéo sau khi bệnh tiến triển và có tổng cộng 41 bệnh nhân nhánh dùng Stivarga phối hợp BSC tiếp tục được điều trị Stivarga sau khi bệnh tiến triển. Các giá trị trung vị của PFS (xác định theo đánh giá của nghiên cứu viên) lần lượt là là 5.0 và 4.5 tháng.
Ung thư biểu mô tế bào gan (HCC)
Hiệu quả lâm sàng và an toàn của Stivarga đã được đánh giá trong một nghiên cứu pha III đa quốc gia, đa trung tâm, ngẫu nhiên, mù đôi, đối chứng giả dược (RESORCE) ở những bệnh nhân bị ung thư biểu mô tế bào gan đã được điều trị trước đó với sorafenib.
Tiêu chí đánh giá hiệu quả chính là Thời gian sống còn toàn bộ (OS). Các tiêu chí phụ là Thời gian sống còn không bệnh tiến triển (Progression-Free Survival - PFS), thời gian đến khi bệnh tiến triển (Time To Progression - TTP), tỷ lệ đáp ứng khối u khách quan (Objective Tumor Response Rate - ORR) và tỷ lệ kiểm soát bệnh (Disease Control Rate - DCR).
Tổng cộng có 573 bệnh nhân HCC được chia ngẫu nhiên theo tỷ lệ 2:1 để nhận hoặc 160 mg regorafenib uống một lần mỗi ngày (n = 379) cùng với BSC (Chăm sóc hỗ trợ tốt nhất-Best Supportive Care) hoặc giả dược (n = 194) cộng với BSC trong 3 tuần điều trị và sau đó là nghỉ điều trị 1 tuần. Liều hàng ngày trung bình đã dùng của regorafenib là 144 mg. Bệnh nhân được lựa chọn để tham gia vào nghiên cứu nếu họ đã bị bệnh tiến triển được xác định trên X quang trong quá trình điều trị bằng sorafenib và nếu họ có tình trạng chức năng gan ở giai đoạn Child-Pugh A. Những bệnh nhân ngừng sử dụng sorafenib kéo dài do độc tính của sorafenib hoặc những người dung nạp ít hơn 400mg sorafenib một lần mỗi ngày trước khi ngừng thuốc, bị loại trừ khỏi nghiên cứu. Phân ngẫu nhiên được thực hiện trong vòng 10 tuần sau khi điều trị với sorafenib lần cuối. Bệnh nhân tiếp tục điều trị bằng Stivarga cho đến khi bệnh tiến triển xác định trên lâm sàng hoặc X quang hoặc có độc tính không chấp nhận được. Tuy nhiên, bệnh nhân có thể tiếp tục điều trị Stivarga sau khi bệnh tiến triển theo quyết định của nghiên cứu viên.
Các đặc điểm về nhân khẩu học và về bệnh lý trước khi điều trị là tương tự nhau giữa nhóm điều trị Stivarga và nhóm dùng giả dược và được trình bày dưới đây cho tất cả 573 bệnh nhân đã được phân ngẫu nhiên:
• Tuổi trung vị: 63 năm
• Nam giới: 88%
• Bệnh nhân da trắng: 36%, châu Á: 41%
• Chỉ số toàn trạng ECOG 0: 66%, chỉ số toàn trạng ECOG 1: 34%
• Child-Pugh A: 98%, Child-Pugh B: 2%
• Bệnh sinh bao gồm Viêm gan B (38%), Viêm gan C (21%), Viêm gan nhiễm mỡ không do rượu (Non-Alcoholic Steato Hepatitis -NASH, 7%)
• Không có xâm lấn mạch máu lớn và khối u lan ngoài gan: 19%
• Giai đoạn B theo phân loại của Hội ung thư gan lâm sàng Barcelona (BCLC): 13%; giai đoạn C theo phân loại BCLC: 87%
• Các thủ thuật gây thuyên tắc mạch hoặc truyền hóa chất qua động mạch tại chỗ-vùng gan: 61%
• Xạ trị trước khi điều trị bằng regorafenib: 15%
• Trung vị thời gian điều trị sorafenib: 7.8 tháng
Việc phối hợp thêm Stivarga với BSC đã cho kết quả thời gian sống còn toàn bộ tốt hơn đáng kể so với nhóm giả dược phối hợp với BSC, với tỷ số nguy cơ là 0,624 [khoảng tin cậy 95% 0,498, 0,782], p = 0,000020 với kiểm định log rank phân tầng, và trung vị OS là 10,6 tháng so với 7,8 tháng (xem Bảng 7 và Hình 4).
Bảng 7: Kết quả về hiệu quả từ nghiên cứu RESORCE
|
Các thông số về hiệu quả |
Tỷ số nguy cơ (Hazard Ratio)* (khoảng tin cậy 95%) |
Giá trị P (một phía) |
Trung vị (95% CI) |
|
|
Stivarga + BSC§ (N=379) |
Giả dược + BSC§ (N=194) |
|||
|
Thời gian sống còn toàn bộ
|
0.627 (0.500,0.785) |
0.000020 |
10.6 tháng (9.1, 12.1) |
7.8 tháng (6.3, 8.8) |
|
Thời gian sống còn không bệnh tiến triển ** |
0.455 (0.371, 0.558) |
<0.000001 |
3.1 tháng (2.8, 4.2) |
1.5 tháng (1.4, 1.6) |
|
Thời gian đến khi bệnh tiến triển** |
0.442 (0.358,0.545) |
<0.000001 |
3.2 tháng (2.9, 4.2) |
1.5 tháng (1.4, 1.6) |
§ Chăm sóc hỗ trợ tốt nhất (Best Supportive Care –BSC)
* Tỷ số nguy cơ Hazard ratio < 1 tốt hơn cho Stivarga
** dựa trên đánh giá của nghiên cứu viên về đáp ứng khối u bằng thang RECIST sửa đổi
Hình 4: Đường cong Kaplan Meier của Thời gian sống còn toàn bộ

Hình 5: Đường cong Kaplan Meier của Thời gian sống còn không bệnh tiến triển (mRECIST)

Các đặc tính dược động học
• Hấp thu
Regorafenib đạt đỉnh nồng độ trong huyết tương trung bình khoảng 2,5 mg/L ở khoảng 3 đến 4 giờ sau khi uống liều duy nhất 160 regorafenib dưới dạng 4 viên mỗi viên có chứa 40 mg. Sinh khả dụng tương đối trung bình của viên nén so với dạng dung dịch uống là 69-83%.
Nồng độ regorafenib và chất chuyển hóa có hoạt tính dược lý của nó M-2 (N-oxide) và M-5 (N-oxide và N- desmethyl) cao nhất khi được uống sau một bữa ăn sáng ít chất béo so với uống sau một bữa ăn sáng nhiều chất béo hoặc uống lúc đói. Mức độ phơi nhiễm với regorafenib tăng 48% khi dùng chung với bữa ăn sáng nhiều chất béo, và 36% khi dùng chung với một bữa ăn sáng ít chất béo, so với ăn chay. Mức phơi nhiễm của chất chuyển hóa M-2 và M-5 là cao hơn khi regorafenib được dùng với bữa ăn sáng ít chất béo so với ăn chay, và thấp hơn khi được dùng với một bữa ăn giàu chất béo so với lúc đói.
• Phân bố
Số liệu nồng độ- thời gian trong huyết tương của regorafenib cũng như các chất chuyển hóa chính cho thấy có nhiều đỉnh trong khoảng thời gian đưa liều 24 giờ, có sự tham gia của tuần hoàn gan-ruột. In vitro, regorafenib gắn với protein huyết tương người với tỷ lệ cao (99.5%).
• Chuyển hóa/chuyển dạng sinh học
Regorafenib được chuyển hóa chủ yếu ở gan bởi chuyển hóa oxy hóa qua trung gian CYP3A4, cũng như được gắn glucuronid qua trung gian UGT1A9. Hai chất chuyển hóa chính và sáu chất chuyển hóa phụ của regorafenib đã được xác định trong huyết tương. Các chất chuyển hóa lưu hành chính của regorafenib trong huyết tương của người là M-2 (N-oxide) và M-5 (N-oxide và N-desmethyl), có hoạt tính dược lý và có nồng độ tương tự như regorafenib ở trạng thái ổn định.
Sự gắn kết protein trên in vitro của M-2 và M-5 cao hơn (theo thứ tự là 99.8% và 99.95%,) so với của regorafenib.
Các chất chuyển hóa có thể bị khử hoặc thủy phân trong đường tiêu hóa bởi hệ vi khuẩn chí, cho phép tái hấp thu thuốc và các chất chuyển hóa dạng không liên hợp (tuần hoàn gan - ruột). Dùng đồng thời một liều duy nhất regorafenib sau khi đã điều trị trước đó với neomycin (là kháng sinh hấp thu kém có tác dụng diệt trừ hệ vi khuẩn chí đường tiêu hóa) không có ảnh hưởng đáng kể đến sự tiếp xúc của regorafenib. Có sự giảm mức độ tiếp xúc của M-2 và M-5 lần lượt là 76% và 86%.
• Thải trừ
Sau khi uống, thời gian bán thải trung bình của regorafenib và chất chuyển hóa M-2 của nó trong huyết tương dao động trong khoảng từ 20 đến 30 giờ trong các nghiên cứu khác nhau. Thời gian bán thải trung bình của chất chuyển hóa M-5 là khoảng 60 giờ (từ 40 đến 100 giờ).
Khoảng 90% liều có hoạt tính phóng xạ đã được tìm thấy trong vòng 12 ngày sau khi dùng thuốc, với khoảng 71% liều được bài tiết trong phân (47% là chất mẹ, 24% là chất chuyển hóa), và khoảng 19% liều được bài tiết trong nước tiểu dưới dạng liên hợp glucuronide.
Sự bài tiết qua nước tiểu của các chất chuyển hóa liên hợp glucuronide giảm dưới 10% ở điều kiện trạng thái ổn định. Hợp chất mẹ tìm thấy trong phân có thể được bắt nguồn từ sự thoái giáng thuốc không được hấp thu trong đường ruột của các chất liên hợp glucuronide hoặc chất chuyển hóa M-2, cũng như của thuốc không được hấp thu.
• Tuyến tính / phi tuyến tính
Sự phơi nhiễm hệ thống của regorafenib ở trạng thái ổn định tăng tỷ lệ với liều khi tăng liều lên đến 60 mg và tăng ít hơn theo tỷ lệ ở liều lớn hơn 60 mg. Tích lũy của regorafenib ở trạng thái ổn định dẫn đến tăng khoảng 2 lần nồng độ trong huyết tương, phù hợp với thời gian bán thải và số lần dùng thuốc. Ở trạng thái ổn định, regorafenib đạt được nồng độ đỉnh huyết tương trung bình khoảng 3,9 mg/L (8,1 micromolar) sau khi uống 160 mg regorafenib và tỷ lệ đỉnh-đáy của nồng độ trong huyết tương trung bình ít hơn 2.
Cả hai chất chuyển hóa, M-2 và M-5, đều thể hiện tính tích lũy phi tuyến tính. Trong khi đó, nồng độ trong huyết tương của M-2 và M-5 sau một liều regorafenib duy nhất thấp hơn nhiều so với nồng độ huyết tương của hợp chất mẹ, nồng độ trong huyết tương trạng thái ổn định của M-2 và M-5 tương đương với nồng độ huyết tương regorafenib.
Thông tin bổ sung về quần thể bệnh nhân đặc biệt
• Bệnh nhân suy gan
Sự phơi nhiễm của regorafenib và các chất chuyển hóa của nó M-2 và M-5 là tương đương trên những bệnh nhân bị suy gan nhẹ (Child-Pugh A) và những bệnh nhân có chức năng gan bình thường. Không có dữ liệu cho các bệnh nhân suy gan Child-Pugh C (nặng). Regorafenib chủ yếu đào thải qua gan, và phơi nhiễm có thể tăng lên trong nhóm dân số bệnh nhân này.
• Bệnh nhân suy thận
Hiện đã có các dữ liệu lâm sàng và dược động học dựa trên mô hình sinh lý cho thấy sự phơi nhiễm với trạng thái ổn định của regorafenib và các chất chuyển hóa của nó M-2 và M-5 là tương tự nhau ở bệnh nhân suy thận nhẹ, trung bình và nặng so với những bệnh nhân có chức năng thận bình thường.
Dược động học của regorafenib chưa được nghiên cứu ở những bệnh nhân bệnh thận giai đoạn cuối. Tuy nhiên, các mô hình dược động học dựa trên sinh lý học không dự đoán bất kỳ sự thay đổi nào liên quan tiếp xúc ở những bệnh nhân này.
• Bệnh nhân lớn tuổi
Tuổi không ảnh hưởng đến dược động học của regorafenib trong khoảng tuổi nghiên cứu (29 - 85 năm).
• Giới tính
Dược động học của regorafenib không bị ảnh hưởng bởi giới tính.
• Khác biệt về dân tộc
Sự phơi nhiễm với regorafenib trong nhiều nhóm dân số châu Á khác nhau (Trung Quốc, Nhật Bản, Hàn Quốc) có cùng khoảng phơi nhiễm với các bệnh nhân da trắng.
• Sinh lý điện tim / kéo dài khoảng QT
Không quan sát thấy có tác dụng kéo dài khoảng QTc sau khi uống regorafenib 160mg ở trạng thái ổn định trong một nghiên cứu QT chuyên biệt ở bệnh nhân ung thư nam và nữ.
Dữ liệu an toàn tiền lâm sàng
• Độc tính toàn thân
Sau khi dùng liều lặp lại cho chuột nhắt, chuột cống và chó, tác dụng có hại đã được quan sát trong một số cơ quan, chủ yếu ở thận, gan, đường tiêu hóa, tim, tuyến giáp, hệ thống tạo máu-/lympho, hệ nội tiết, hệ sinh sản và da. Các tác dụng này xảy ra khi phơi nhiễm hệ thống trong khoảng hoặc thấp hơn sự phơi nhiễm được dự đoán trên người (dựa trên so sánh AUC).
Sự thay đổi của răng và xương và các tác dụng ngoại ý lên hệ sinh sản đã được thông báo thấy ở chuột non và các động vật cũng như chuột đang tăng trưởng và chỉ ra một nguy cơ tiềm ẩn cho trẻ em và thanh thiếu niên.
• Độc tính trên gen và khả năng sinh ung thư
Các nghiên cứu về khả năng gây ung thư của regorafenib chưa được thực hiện.
Không có chỉ định để thử nghiệm khả năng gây độc tính trên gen của regorafenib trong các thử nghiệm tiêu chuẩn in vitro và in vivo ở chuột.
• Độc tính trên sinh sản và phát triển
Các nghiên cứu chuyên biệt trên sinh sản chưa được thực hiện. Tuy nhiên, tiềm năng của regorafenib ảnh hưởng xấu đến sinh sản nam và nữ đã được xem xét dựa trên các thay đổi hình thái học trong tinh hoàn, buồng trứng và tử cung quan sát thấy sau khi dùng thuốc với liều lặp lại ở chuột và chó với mức phơi nhiễm nhỏ hơn mức phơi nhiễm dự kiến ở người (dựa trên so sánh AUC). Những thay đổi chỉ hồi phục một phần.
Một tác dụng của regorafenib trên sự phát triển trong tử cung đã được thể hiện trên thỏ ở mức phơi nhiễm nhỏ hơn mức phơi nhiễm dự kiến ở người (dựa trên so sánh AUC). Những phát hiện chính bao gồm dị tật ở hệ tiết niệu, tim và các mạch máu lớn, và xương
Không
36 tháng kể từ ngày sản xuất
Giữ lọ đậy kín sau khi mở lần đầu tiên. Sau khi mở nắp chai, thuốc đã thể hiện tính ổn định trong 7 tuần. Sau đó, sản phẩm điều trị cần được loại bỏ.
Không quá 30oC
Lưu trữ trong bao bì ban đầu để tránh ẩm.
Để chất hút ẩm trong chai.
Lọ 28 viên nén. Hộp 1 lọ hoặc 3 lọ.
Ấn nắp đậy theo hướng dẫn trên nắp đồng thời xoay sang bên trái. Giữ lọ đậy kín sau khi mở lần đầu tiên. Không được uống viên nang hút ẩm.
Bất kỳ sản phẩm thuốc không sử dụng hoặc vật liệu phế thải cần được xử lý phù hợp với yêu cầu của cơ sở điều trị
Để thuốc xa tầm với của trẻ em
Đọc kỹ hướng dẫn trước khi sử dụng
Để biết thêm thông tin, xin vui lòng hỏi lời khuyên của bác sĩ
Nhà sản xuất:
Bayer AG
Kaiser-Wilhelm-Allee
51368 Leverkusen, Đức
Ngày phê duyệt tài liệu: 02/08/2018
Stivarga/EU SmPC/220617/PI VN01
